Research projects


Phonon anharmonicity in crystals
Lattice anharmonicity plays a critical role in determining important thermodynamics properties such as thermal conductivity and thermal expansion. From the microscopic point of view, the lattice anharmonicity can be described by the deviation of the phonon frequencies with respect the harmonic ones (frequency shift) and its uncertainty around this value (linewidths). In highly anharmonic systems, these phonon anharmonic properties can be calculated at arbitrary precision from molecular dynamics (MD) simulations but it becomes extremely complex to do it manually. For this purpose, I developed, implemented and tested DynaPhoPy (https://abelcarreras.github.io/DynaPhoPy/), an open-source software to calculate the anharmonic properties of crystals from MD calculations [Comput Phys Commun, 2017, 221, 221–34]. This code is fully compatible with phonopy, one of the most popular codes for phonon calculations based on lattice dynamics approximation, which is used to determine the harmonic phonon eigenvectors needed as input for DynaPhoPy.
When calculated from first principles, MD simulations are usually computationally very expensive. To circumvent this issue, many large and complex systems are studied using empirical potentials that drastically reduce the computational cost. LAMMPS is one of the most popular software to perform classical MD simulations using empirical potentials. To use LAMMPS MD simulations with DynaPhoPy, I developed phonoLAMMPS (https://github.com/abelcarreras/phonolammps), a software that provides an interface between LAMMPS and phonopy and allows to compute the phonon eigenvectors required by DynPhoPy.
Automation of quantum chemistry calculations
PyQchem (https://github.com/abelcarreras/PyQchem) is an open-source python interface for Q-Chem designed to automize complex workflows using Q-Chem with other software. My choice of python as language for this implementation has been driven by its extensive adoption within the scientific community during the last decade owing to its flexibility and simplicity. PyQchem is designed to create inputs, launch calculations and parse the outputs seamlessly from python scripts. It allows the user to easily and efficiently perform usual tasks such as benchmarks, scans, reaction paths, and other procedures that require multiple Q-Chem calculations. It also provides tools for post-processing electronic structure calculations and compute key properties of molecular systems.
PyQchem is designed with a modular structure (Fig. 1) that allows straightforward implementation of new tools that make use of electronic structure calculations. In the following list, I present a selection of the currently available tools and workflows included in the PyQchem package:
• Continuous Symmetry analysis of the orbitals and wave functions
• Orbital classification according to symmetry criteria
• Automated diabatization scheme for RASCI and TDDFT
• Excited states symmetry analysis
• G-tensor calculation for RASCI states
• Mono-electronic and bi-electronic integrals extraction
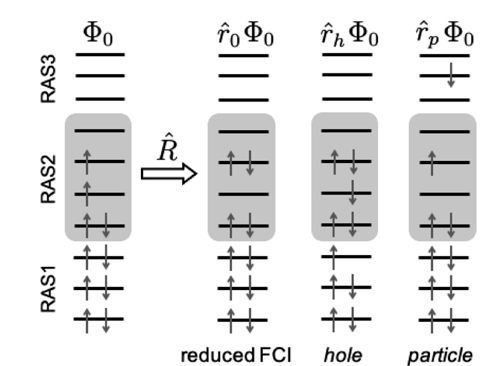
• Split RASCI calculation
• Vibrational spectra simulation
Electronic structure methods for excited states
The study of excited states of molecules is crucial to understand light-matter interaction phenomena such as photon absorption, energy transport, fluorescence or phosphorescence (amongst others), which have many applications in modern technology. These include organic thin-film-transistors, light-emitting diodes, solar cells, sensors, and many others. To describe these phenomena, a large number of different methodologies have been developed within the quantum chemistry field.
In this project I developed tools for implementing the Restricted Active Space Configuration Interaction (RASCI) method in the Q-Chem software suite. This electronic structure approach enables the efficient description of correlated ground and low-lying excited states with a moderate computational cost. These contributions have been made in my capacity as an official Q-Chem developer [J Chem Phys, 2021, 155, 084801]. Below is a representative, though incomplete, list of my most recent implementations in Q-Chem:
• Orbital localization scheme for RASCI
• Spin polarization for Short-range DFT implementation for restricted reference configurations
• Mulliken analysis of the electronic and transition density of excited states.
• Diabatization scheme of electronic excited states using most popular methods such as Boys, Edmiston-Ruedenberg and Dipole-Quadrupole
• Symmetry analysis of excited states computed with RASCI method
• Transition Natural Orbitals implementation for RASCI
• Spin-Orbit Coupling for RASCI states
• Fractional electronic density and indices
Development of symmetry tools
Continuous symmetry measures (CSMs) provide a quantitative evaluation of how much a structure deviates from a specific point symmetry. Since my PhD, advancing the development and application of CSMs has been a central focus of my research. Within this framework, I have created a suite of computational tools designed to analyze the symmetry properties of a wide range of chemical objects in great detail, enabling deeper insights into their structural and functional characteristics:
Cubesym (https://github.com/abelcarreras/cubesym):
a code to calculate the CSM of the electronic density or molecular orbitals using a numerical integration approach from a GAUSSIAN cube formatted file. I used this software to characterize the sandwich and piano-stool complexes along the highest order symmetry axis [Chem - A Eur J, 2011, 17, 359–67].
Symgroup (https://github.com/abelcarreras/symgroup):
a FORTRAN code for calculating the CSM of molecular structures. I also have implemented a python interface and other compatibility updates.
WFNSYM (https://github.com/abelcarreras/WFNSYM):
this is a code for calculating the CSM of the wavefunction of molecules [J Comput Chem, 2013, 34, 1321–31]. WFNSYM has important applications in the automation of sophisticated quantum mechanics methods that rely on symmetry, such as coupled cluster (CC) and configuration interaction (CI), to improve their performance and analyze their results. I implemented a python interface for this code that I am currently maintaining. I also have implemented several improvements, notably adding compatibility with multiple quantum chemistry codes.
Cosymlib
(https://github.com/GrupEstructuraElectronicaSimetria/cosymlib): since 2017, I have led a project to integrate all the CSM codes developed in FORTRAN into a python library. This library aims to be a reference in the theoretical chemistry community for CSM analysis, grouping old FORTRAN codes under a common modern interface. The implementation of this library was the central PhD project of Efrem Bernuz (2017-2021) who successfully graduated in 2021 under my co-supervision. This library has been used in several studies, e.g., [Chem Eur J 2019, 25, 673–691] and [J Mater Chem C, 2022, 10, 13826-13833].
PoSym (https://github.com/abelcarreras/posym):
I developed this Python library to analyze the symmetry of a wide range of chemical objects, including wave functions, electronic densities, molecular orbitals, normal modes, and more. Designed for versatility, the library can be seamlessly integrated as a module into other Python codes requiring symmetry analysis. I plan to incorporate it into Cosymlib, enhancing its features and adding new functionality.
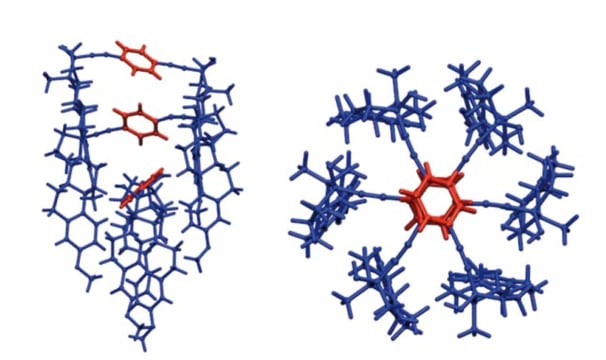
Simulation and characterization of molecular rotors
Molecular rotors are molecular systems that, analogously to macroscopic rotors, present an essentially rigid structure with only a few rotational degrees of freedom. The dynamical study of these compounds requires long simulations in order to observe correlated gearing phenomena. To archive this with a good degree of precision in a reasonable period of time, it was necessary to develop a model in which those systems are treated as an assembly of rigid fragments bound by flexible hinge-like structures. I developed a methodology based on two FORTRAN codes, ACODYM and ESMOCA, able to simulate the properties of molecular rotors from molecular dynamics and classical Monte Carlo simulations, respectively. These codes have used to study large and flexible molecular rotors [Phys Chem Chem Phys, 2014, 16, 12980-12986] and [Phys Chem Chem Phys, 2019, 21, 11395-11404].
Exciton dynamics in optoelectronic materials
Organic semiconductors have been studied extensively in recent years for their applications in optoelectronic materials. The special characteristic of these materials lies in their ability to collect, transfer and transport localized excited states, known as excitons, through the material with high control, which makes them suitable for applications in electronics like organic thin-film-transistors, light-emitting diodes, solar cells, sensors, and many others.
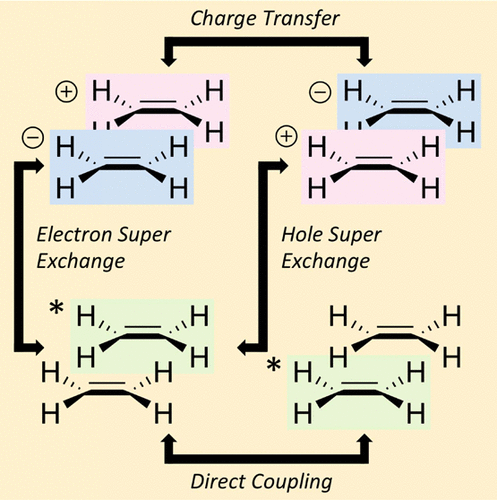
The exhaustive description of these phenomena requires taking into account a large variety of processes related to exciton creation, diffusion, charge generation/recombination and decay. To simulate exciton dynamics in these materials, [C8] I developed Kimonet (https://github.com/abelcarreras/kimonet) a software using electronic structure calculations and based on Kinetic Monte Carlo (kMC).
The central point of kMC is the determination of individual transfer rates for each of the processes, which can be modelled by the Fermi’s golden rule. In this approach, the electronic and vibrational couplings are treated as independent variables. However, in some cases, particularly due the symmetry of the system, the computed electronic coupling can be zero while the electronic-vibrational coupling be important leading to non-zero transfer rates. We explored this possibility for a simple model of singlet exciton diffusion on naphthalene crystal [J Chem Phys, 2022, 156, 044112]. For this study, [C9] I employed GromorG (https://github.com/abelcarreras/gromorg), a python interface for GROMMACS I developed to simulate molecular dynamics of organic crystals.
I have also explored the effect of the electronic coupling on the dynamics of thermally activated delayed fluorescence (TADF) process. [ChemPhotoChem, 2022, 6, e202200066]. In this process, molecular species in a non-emitting excited state (triplet state) can incorporate surrounding thermal energy to access the singlet manifold and undergo light emission. In this work, we modeled and rationalized the different mechanisms involved in TADF as a function of several parameters such as the relative position of relevant close-lying excited states.
Quantum algorithms for quantum chemistry
Quantum simulation of chemical systems stands out as a promising application for quantum computers in the near term. These computers promise to speed up the calculations with respect to classical computers making use of two quantum properties: quantum parallelization and efficient encoding. In this context, variational quantum eigensolvers (VQE) are one of the most promising family of algorithms. VQE are a type of hybrid algorithms in which part of the calculation is done by a quantum computer (Hamiltonian evaluation) and another by a classical computer (parameter optimization).
I developed multiVQE, a Python library implementing several of the most promising state-of-the-art variational quantum eigensolver (VQE) algorithms for solving molecular electronic Hamiltonians. These include standard VQE, ADAPT-VQE, and qubit-ADAPT-VQE. Furthermore, I have contributed to advancing VQE methods for chemistry applications, as demonstrated in [J Chem Theory Comput, 2024, 12, 5133]. This work introduced physically inspired improvements to the ADAPT-VQE algorithm, such as the use of correlated orbitals and basis projections, enhancing its efficiency and accuracy.
In addition, I am collaborating with Dr. Rodríguez (BasQ) and IBM Quantum on developing a quantum Lanczos algorithm to compute the ground state of electronic Hamiltonians, further expanding the toolkit for quantum chemistry applications.